02_01Lithium_dimer¶
00 Introduction¶
From this tutorial, you can learn how to calculate all-electron Variational Monte Carlo (VMC) and lattice regularized diffusion Monte Carlo (LRDMC) energies of the Li2 dimer with various ansatz. JDFT, JSD, JsAGPs, JAGPu, and JAGP(JPf).
Reference values (https://aip.scitation.org/doi/suppl/10.1063/1.3288054)
- Li atom
HF = -7.4327 Ha
Exact = -7.4781 Ha
- Li2 dimer
dbond = 2.67330 angstrom (J. Chem. Phys. 129 204105)
HF = -14.8715 Ha
Exact = -14.9951 Ha
Ebond = 24.4 kcal/mol = 38.9 mHa
01 Li2 dimer and Li atom - JDFT ansatz¶
01-01 Li2 dimer: Preparing a wave function¶
Prepare Li2 dimer structure, Li2.xyz
2
comment line
Li 0.0000 0.0000 -2.52589975576638451762
Li 0.0000 0.0000 2.52589975576638451762
and prepare makefort10.input and convertfort10mol.input and
# makefort10.input
&system
posunits='bohr'
natoms=2
ntyp=1
pbcfort10=.false.
/
&electrons
twobody=-15
twobodypar=1.00
onebodypar=1.00
no_4body_jas=.false.
neldiff=0
/
&symmetries
eqatoms=.true.
rot_det=.true.
symmagp=.true.
/
ATOMIC_POSITIONS
3.0000000000000000 3.0000000000000000 0.0000000000000000 0.0000000000000000 -2.5259024244810400
3.0000000000000000 3.0000000000000000 0.0000000000000000 0.0000000000000000 2.5259024244810400
/
ATOM_3
&shells
nshelldet=11
nshelljas=5
/
1 1 16
1 14.24
1 1 16
1 4.581
1 1 16
1 1.58
1 1 16
1 0.564
1 1 16
1 0.07345
1 1 16
1 0.02805
1 1 36
1 1.534
1 1 36
1 0.2749
1 1 36
1 0.07362
1 1 36
1 0.02403
1 1 68
1 0.1144
# Parameters atomic Jastrow wf
1 1 16
1 4.581
1 1 16
1 1.58
1 1 16
1 0.564
1 1 36
1 0.2749
1 1 36
1 0.07362
# convertfort10mol.input
&control
epsdgm=-1d-14
/
&mesh_info
ax=0.20
nx=64
ny=68
nz=128
/
&molec_info
nmol=3
nmolmax=3
nmolmin=3
/
You may obtain makefort10.input. Next, you can run makefort10.sh:
kosukenoMBP% ./makefort10.sh
where,
# makefort10.sh
kosukenoMBP% cat makefort10.sh
makefort10.x < makefort10.input > out_make
mv fort.10_new fort.10
mv fort.10 fort.10_in
convertfort10mol.x < convertfort10mol.input > out_mol
mv fort.10_new fort.10
The generated wave function fort.10 is a symmetric Jastrow Antisymmetrized Geminal Power (JsAGPs).
01-02 Li2 dimer: generate a trial wave function using DFT.¶
The next step is to generate a trial wave function using the built-in DFT code. This is the minimal input file:
# prep.input
&simulation
itestr4=-4
iopt=1
double_mesh=.true
/
&pseudo
/
&vmc
/
&optimization
molopt=1
/
&readio
/
¶meters
/
&molecul
ax=0.2
ay=0.2
az=0.2
nx=64
ny=64
nz=128
/
&dft
maxit=50
epsdft=1d-5
mixing=1.0d0
typedft=1
optocc=0
nelocc=3
l0_at=0.8320335292207617
scale_z=5
scale_hartree=-1.00
corr_hartree=.true.
linear=.false.
/
2 2 2
Then, you can run DFT by typing:
prep-serial.x < prep.input > out_prep
You can optimize one-body Jastrow.
#onebody-script for bash on mac
b_onebody_list="0.5 0.6 0.7 0.8 0.9 1.0 1.1 1.2 1.3 1.4 1.5"
root_dir=`pwd`
for b_onebody in $b_onebody_list
do
mkdir onebody_$b_onebody
cd onebody_$b_onebody
cp $root_dir/temp/fort.10 ./
cp $root_dir/temp/prep.input ./
gsed -i -e "s/b_onebody/$b_onebody/g" fort.10
prep-serial.x < prep.input > out_prep
cd $root_dir
done
echo "b_onebody dft energy" > result.out
for b_onebody in $b_onebody_list
do
cd onebody_$b_onebody
energy=`grep "Final self consistent energy" ./out_prep | awk '{print $7}'`
echo "${b_onebody} ${energy}" >> ../result.out
cd $root_dir
done
You may get
kosukenoMBP% cat result.out
b_onebody dft energy
0.5 -14.740030455469444
0.6 -14.746632760686529
0.7 -14.749842736580408
0.8 -14.750834382315727
0.9 -14.750935229881659 <- the lowest one
1.0 -14.750763468915963
1.1 -14.750537472297879
1.2 -14.750319868767475
1.3 -14.750124626530374
1.4 -14.749953844050875
1.5 -14.749807044323163
Here, the DFT double-grid integration scheme is employed.
l0_at The radius where the double-grid is used (Bohr)
scale_z The number of grids used for the double grid. Indeed, the grid sizes in the double-grid region are ax/scale_z, ay/scale_z, and ax/scale_z
scale_hartree, corr_hartree, and linear can be default values.
01-03 Li2 dimer: Jastrow factor optimization (WF=JDFT)¶
One should refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd 02optimization/
cp ../01trial_wavefunction/02onebody_jastrow_opt/onebody_0.9/fort.10_new fort.10
cp fort.10 fort.10_dft
mpirun -np 4 turborvb-mpi.x < datasmin.input > out_min &
rm -r turborvb.scratch/
plot_Energy.sh out_min
plot_devmax.sh out_min
readalles.x << ____EOS
1 41 10
0
1000
____EOS
01-04 Li2 dimer: VMC (WF=JDFT)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd 03vmc/
cp ../02optimization/fort.10 ./
cp ../02optimization/datasmin.input ave.in
gsed -i -e 's/ngen=.*/ngen=1/g' ave.in
gsed -i -e 's/iopt=.*/iopt=1/g' ave.in
line_num_key=`grep "unconstrained" -n fort.10 | cut -d ":" -f 1`
line_num=`expr $line_num_key + 1`
line=`gsed -n ${line_num}p fort.10`
mod_line=`echo $line | gsed -e 's/0$/ 1/'`
gsed -i "${line_num}d" fort.10
gsed -i "${line_num_key}a $mod_line" fort.10
turborvb-serial.x < ave.in > out_ave
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
forcevmc.sh 20 5 1
cat pip0.d | grep "Energy"
...
number of bins read = 14996
Energy = -14.9757876300445 4.853630781726438E-004
Variance square = 7.979609547808308E-002 2.379635260709342E-004
Est. energy error bar = 4.748991835467470E-004 2.848268581908139E-006
Est. corr. time = 3.39078657629560 3.817021016290687E-002
VMC (JDFT) = -14.9759(9) Ha
01-05 Li2 dimer: LRDMC (WF=JDFT)¶
One should refer to the Hydrogen tutorial for the details. Here, only needed explanations and commands are shown.
# datasfn.input
&simulation
itestr4=-6
ngen=200000
iopt=2
/
&pseudo
/
&dmclrdmc
tbra=0.1d0
etry=-15.00d0
alat=-0.20
!alat2=0.0d0
iesrandoma=.true.
/
&readio
/
¶meters
/
Here are brief explanations of the variables for a LRDMC calculation:
&dmclrdmc section
tbra projection time (i.e, \exp(-\tau \cdot \hat{\mathcal{H}})). Set 0.1 in general. However, for a heavy element, it is better to choose a smaller value. Please check Average number of survived walkers in out_fn
Av. num. of survived walkers/ # walkers in the branching
0.9939
if the number is too small, try smaller tbra.
etry Put a DFT of VMC energy. \Gamma in eq.6 of the review paper is set 2 \times etry
alat The lattice space for discretizing the Hamitonian. If you do a single grid calculation (i.e., alat2=0.0d0), please put a negative value. If you do a double-grid calculation (See. Nakano’s paper), put a positive value and set iesrandoma=.true.. This trick is needed for satisfying the detailed-valance condition.
alat2 The corser lattice space used in the double-grid calculation. If you put 0.0d0, TurboRVB does a single grid calculation. If you want to do a double-grid calculation for a compound include Z > 2 element, please comment out alat2 because alat2 is automatically set (See Nakano’s paper).
Prepare different working directories, copy fort.10 to each directory, and set the corresponding alat.
alat_0.8Z
alat_1.0Z
alat_1.2Z
alat_1.5Z
And then, run each LRDMC calculation after generating initial electron configurations at the VMC level.
cd ../03lrdmc/
cp ../02vmc/fort.10 .
alat_list="0.8Z 1.0Z 1.2Z 1.5Z"
lrdmc_root_dir=`pwd`
for alat in $alat_list
do
cd alat_${alat}
cp ${lrdmc_root_dir}/fort.10 ./fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
mpirun -np 4 turborvb-mpi.x < datasfn.input > out_fn;
cd ${lrdmc_root_dir}
done
num=0
echo -n > ${lrdmc_root_dir}/evsa.gnu
for alat in $alat_list
do
cd alat_${alat}
num=`expr ${num} + 1`
echo "10 20 5 1" | readf.x
alat_d=`grep alat= datasfn.input | cut -f 2 -d '='`
echo -n "${alat_d} " >> ${lrdmc_root_dir}/evsa.gnu
tail -n 1 fort.20 | awk '{print $1, $2}' >> ${lrdmc_root_dir}/evsa.gnu
cd ${lrdmc_root_dir}
done
gsed "1i 2 ${num} 4 1" evsa.gnu > evsa.in
One has collected all LRDMC energis into evsa.in
# Z=3 (Li)
2 4 4 1
0.22222 -14.9907899448815 3.101461151525789E-004 <- 1/(1.5*Z)
0.27778 -14.9920193657668 3.175522770991489E-004 <- 1/(1.2*Z)
0.33333 -14.9933364338623 3.387085675247969E-004 <- 1/(1.0*Z)
0.41667 -14.9969823559963 3.650299072111256E-004 <- 1/(0.8*Z)
funvsa.x is a tool for a quadratic fitting:
funvsa.x < evsa.in > evsa.out
You can see
Reduced chi^2 = 0.258290367368527
Coefficient found
1 -14.9894036676827 1.126607950073031E-003 <- E_0
2 -2.333404434142712E-002 2.310746587271902E-002 <- k_1
3 -0.116381701497219 0.102032433967512 <- k_2
- Li2 dimer
HF = -14.8715 Ha
VMC(JDFT) = -14.9759(9) Ha
LRDMC(JDFT) = -14.9894(11) Ha
Exact = -14.9951 Ha
01-06 Li atom: preparation of a wave function¶
and prepare makefort10.input and convertfort10mol.input and
# makefort10.input
&system
posunits='bohr'
natoms=1
ntyp=1
pbcfort10=.false.
/
&electrons
twobody=-15
twobodypar=1.00
onebodypar=0.90
no_4body_jas=.false.
neldiff=1 !! unpaired electrons
/
&symmetries
eqatoms=.true.
rot_det=.true.
symmagp=.true.
/
ATOMIC_POSITIONS
3.0000000000000000 3.0000000000000000 0.0000000000000000 0.0000000000000000 0.0000000000000000
/
ATOM_3
&shells
nshelldet=11
nshelljas=5
/
1 1 16
1 14.24
1 1 16
1 4.581
1 1 16
1 1.58
1 1 16
1 0.564
1 1 16
1 0.07345
1 1 16
1 0.02805
1 1 36
1 1.534
1 1 36
1 0.2749
1 1 36
1 0.07362
1 1 36
1 0.02403
1 1 68
1 0.1144
# Parameters atomic Jastrow wf
1 1 16
1 4.581
1 1 16
1 1.58
1 1 16
1 0.564
1 1 36
1 0.2749
1 1 36
1 0.07362
# convertfort10mol.input
&control
epsdgm=-1d-14
/
&mesh_info
ax=0.20
nx=64
ny=64
nz=64
/
&molec_info
nmol=1
nmolmax=1
nmolmin=1
/
You may obtain makefort10.input. Next, you can run makefort10.sh:
kosukenoMBP% ./makefort10.sh
where,
# makefort10.sh
kosukenoMBP% cat makefort10.sh
makefort10.x < makefort10.input > out_make
mv fort.10_new fort.10
mv fort.10 fort.10_in
convertfort10mol.x < convertfort10mol.input > out_mol
mv fort.10_new fort.10
The generated wave function fort.10 is a symmetric Jastrow Antisymmetrized Geminal Power (JsAGPs).
01-07 Li atom: generate a trial wave function using DFT.¶
One should refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
mpirun -np 4 prep-mpi.x < prep.input > out_prep
01-08 Li atom: Jastrow factor optimization (WF=JDFT)¶
One should refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd 02optimization/
cp ../01trial_wavefunction/01DFT/fort.10_new fort.10
cp fort.10 fort.10_dft
mpirun -np 4 turborvb-mpi.x < datasmin.input > out_min &
rm -r turborvb.scratch/
plot_Energy.sh out_min
plot_devmax.sh out_min
readalles.x << ____EOS
1 41 10
0
1000
____EOS
01-09 Li atom: VMC (WF=JDFT)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd 03vmc/
cp ../02optimization/fort.10 ./
cp ../02optimization/datasmin.input ave.in
gsed -i -e 's/ngen=.*/ngen=1/g' ave.in
gsed -i -e 's/iopt=.*/iopt=1/g' ave.in
line_num_key=`grep "unconstrained" -n fort.10 | cut -d ":" -f 1`
line_num=`expr $line_num_key + 1`
line=`gsed -n ${line_num}p fort.10`
mod_line=`echo $line | gsed -e 's/0$/ 1/'`
gsed -i "${line_num}d" fort.10
gsed -i "${line_num_key}a $mod_line" fort.10
turborvb-serial.x < ave.in > out_ave
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
forcevmc.sh 20 5 1
cat pip0.d
...
number of bins read = 14996
Energy = -7.47539042703452 2.682804200575495E-004
Variance square = 2.847683107759093E-002 1.295272783812896E-004
Est. energy error bar = 2.683664075931440E-004 1.733280980510503E-006
Est. corr. time = 3.03421296313493 3.544802996972372E-002
VMC (JDFT) = -14.9759(9) Ha (Li2 dimer) VMC (JDFT) = -7.4754(3) Ha (Li atom)
- Li2 dimer
Ebond = 38.9 mHa (experiment)
Ebond = 25.1 mHa (VMC-JDFT)
01-10 Li atom: LRDMC (WF=JDFT)¶
One should refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../03lrdmc/
cp ../02vmc/fort.10 .
alat_list="0.8Z 1.0Z 1.2Z 1.5Z"
lrdmc_root_dir=`pwd`
for alat in $alat_list
do
cd alat_${alat}
cp ${lrdmc_root_dir}/fort.10 ./fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
mpirun -np 4 turborvb-mpi.x < datasfn.input > out_fn;
cd ${lrdmc_root_dir}
done
num=0
echo -n > ${lrdmc_root_dir}/evsa.gnu
for alat in $alat_list
do
cd alat_${alat}
num=`expr ${num} + 1`
echo "10 20 5 1" | readf.x
alat_d=`grep alat= datasfn.input | cut -f 2 -d '='`
echo -n "${alat_d} " >> ${lrdmc_root_dir}/evsa.gnu
tail -n 1 fort.20 | awk '{print $1, $2}' >> ${lrdmc_root_dir}/evsa.gnu
cd ${lrdmc_root_dir}
done
gsed "1i 2 ${num} 4 1" evsa.gnu > evsa.in
One has collected all LRDMC energis into evsa.in
# Z=3 (Li)
2 4 4 1
0.22222 -7.47824129633644 1.450042242028465E-004 <- 1/(1.5*Z)
0.27778 -7.47833093539214 1.562945930953174E-004 <- 1/(1.2*Z)
0.33333 -7.47909688312053 1.628394384627272E-004 <- 1/(1.0*Z)
0.41667 -7.48032309244573 1.866069140147947E-004 <- 1/(0.8*Z)
funvsa.x is a tool for a quadratic fitting:
funvsa.x < evsa.in > evsa.out
You can see
Reduced chi^2 = 1.52438473425829
Coefficient found
1 -7.47794022870940 5.344739888993537E-004 <- E_0
2 -1.467218812266460E-003 1.107590136996555E-002 <- k_1
3 -7.145382242986854E-002 4.939621583555785E-002 <- k_2
- Li2 dimer
LRDMC(JDFT) = -14.9894(11) Ha
LRDMC(JDFT) = -7.4779(5) Ha
Ebond = 38.9 mHa (experiment)
Ebond = 25.1 mHa (VMC-JDFT)
Ebond = 33.6 mHa (LRDMC-JDFT)
02 Li2 dimer and Li atom - JSD ansatz¶
02-01 Li2 dimer and Li atom: VMC-optimization (WF=JSD)¶
One can optimize the determinant part of the JDFT ansatz, the resultant
ansatz is called JSD.
In this case, one does not have to convert the ansatz, just put the
following section in datasmin.input at the optimization step.
# datasmin.input for the Li dimer
&optimization
molopt=-1
...
/
¶meters
iesd=1
iesfree=1
iessw=1
/
&molecul
ax=0.10
ay=0.10
az=0.10
nx=150
ny=150
nz=250
nmolmin=3
nmolmax=3
/
# datasmin.input for the Li atom
&optimization
molopt=-1
...
/
¶meters
iesd=1
iesfree=1
iessw=1
/
&molecul
ax=0.10
ay=0.10
az=0.10
nx=150
ny=150
nz=150
nmolmin=1
nmolmax=1
/
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
# Li2 dimer - vmc opt
cp ../../../01JDFT/01Li2_dimer/03vmc/fort.10 .
cp fort.10 fort.10_jdft
mpirun -np 4 turborvb-mpi.x < datasmin.input > out_min;
readalles.x << ____EOS
1 181 1 0
0
1000
____EOS
# Li atom - vmc opt
cp ../../../01JDFT/02Li_atom/03vmc/fort.10 .
cp fort.10 fort.10_jdft
mpirun -np 4 turborvb-mpi.x < datasmin.input > out_min;
readalles.x << ____EOS
1 181 1 0
0
1000
____EOS
02-02 Li2 dimer and Li atom: VMC (WF=JSD)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../02vmc
cp ../01optimization/fort.10 .
cp ../01optimization/datasmin.input ave.in
gsed -i -e 's/ngen=.*/ngen=1/g' ave.in
gsed -i -e 's/iopt=.*/iopt=1/g' ave.in
line_num_key=`grep "unconstrained" -n fort.10 | cut -d ":" -f 1`
line_num=`expr $line_num_key + 1`
line=`gsed -n ${line_num}p fort.10`
mod_line=`echo $line | gsed -e 's/0$/ 1/'`
gsed -i "${line_num}d" fort.10
gsed -i "${line_num_key}a $mod_line" fort.10
turborvb-serial.x < ave.in > out_ave
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
forcevmc.sh 10 2 1
cat pip0.d | grep "Energy"
02-03 Li2 dimer and Li atom: LRDMC (WF=JSD)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../03lrdmc/
cp ../02vmc/fort.10 .
alat_list="0.8Z 1.0Z 1.2Z 1.5Z"
lrdmc_root_dir=`pwd`
for alat in $alat_list
do
cd alat_${alat}
cp ${lrdmc_root_dir}/fort.10 ./fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
mpirun -np 4 turborvb-mpi.x < datasfn.input > out_fn;
cd ${lrdmc_root_dir}
done
num=0
echo -n > ${lrdmc_root_dir}/evsa.gnu
for alat in $alat_list
do
cd alat_${alat}
num=`expr ${num} + 1`
echo "10 20 5 1" | readf.x
alat_d=`grep alat= datasfn.input | cut -f 2 -d '='`
echo -n "${alat_d} " >> ${lrdmc_root_dir}/evsa.gnu
tail -n 1 fort.20 | awk '{print $1, $2}' >> ${lrdmc_root_dir}/evsa.gnu
cd ${lrdmc_root_dir}
done
gsed "1i 2 ${num} 4 1" evsa.gnu > evsa.in
funvsa.x < evsa.in > evsa.out
Finally I got:
Li2 dimer
HF = -14.8715 Ha
VMC(JDFT) = -14.9759(9) Ha
LRDMC(JDFT) = -14.9894(11) Ha
VMC(JSD) = -14.9803(5) Ha
LRDMC(JSD) = -14.9909(12) Ha
Exact = -14.9951 Ha
Li atom
HF = -7.4327 Ha
VMC(JDFT) = -7.4754(3) Ha
LRDMC(JDFT) = -7.4779(5) Ha
VMC(JSD) = -7.4769(2) Ha
LRDMC(JSD) = -7.4779(4) Ha
Exact = -7.4781 Ha
Binding energy
Ebond = 38.9 mHa (experiment)
03 Li2 dimer and Li atom - JsAGPs ansatz¶
The procedure is the almost same as in the Hydrogen-dimer tutorial.
Three hybrid-orbitals (nhyb=2) were employed here.
03-01 Li2 dimer and Li atom: Conversion of WF (WF=JsAGPs)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
# Li2 dimer - conversion
cp ../../../02JSD/01Li2_dimer/02vmc/fort.10 ./fort.10_in
cp ../../../01JDFT/01Li2_dimer/01trial_wavefunction/00makefort10/makefort10.input .
twobodyjas=`grep -A 1 "Parameters Jastrow two body" ./fort.10_in | tail -n -1 | awk '{print $2}'`
onebodyjas=`grep -A 1 "Parameters Jastrow two body" ./fort.10_in | tail -n -1 | awk '{print $3}'`
echo "twobodyjas=${twobodyjas} onebodyjas=${onebodyjas}"
gsed -i -e "/twobodypar/c\ twobodypar=${twobodyjas}" \
-e "/onebodypar/c\ onebodypar=${onebodyjas}" \
makefort10.input
hyb_line=`grep "nshelljas" -n makefort10.input | cut -d ":" -f 1`
hyb_line=`expr $hyb_line + 1`
gsed -i "${hyb_line}i ndet_hyb=2" makefort10.input
makefort10.x < ./makefort10.input > out_make
mv fort.10_new fort.10_out
convertfort10.x < ./convertfort10.input > out_conv
grep "Overlap square Geminal" out_conv
mv fort.10_new fort.10
cp fort.10_in fort.10_new
copyjas.x > out_copyjas
cleanfort10.x > out_cleanfort10
cp fort.10_clean fort.10
# Li atom - conversion
cp ../../../02JSD/02Li_atom/02vmc/fort.10 ./fort.10_in
cp ../../../01JDFT/02Li_atom/01trial_wavefunction/00makefort10/makefort10.input .
twobodyjas=`grep -A 1 "Parameters Jastrow two body" ./fort.10_in | tail -n -1 | awk '{print $2}'`
onebodyjas=`grep -A 1 "Parameters Jastrow two body" ./fort.10_in | tail -n -1 | awk '{print $3}'`
echo "twobodyjas=${twobodyjas} onebodyjas=${onebodyjas}"
gsed -i -e "/twobodypar/c\ twobodypar=${twobodyjas}" \
-e "/onebodypar/c\ onebodypar=${onebodyjas}" \
makefort10.input
hyb_line=`grep "nshelljas" -n makefort10.input | cut -d ":" -f 1`
hyb_line=`expr $hyb_line + 1`
gsed -i "${hyb_line}i ndet_hyb=2" makefort10.input
makefort10.x < ./makefort10.input > out_make
mv fort.10_new fort.10_out
convertfort10.x < ./convertfort10.input > out_conv
grep "Overlap square Geminal" out_conv
mv fort.10_new fort.10
cp fort.10_in fort.10_new
copyjas.x > out_copyjas
cleanfort10.x > out_cleanfort10
cp fort.10_clean fort.10
03-02 Li2 dimer and Li atom: conversion check (WF=JsAGPs)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../02conversion_check/
cp ../01convert_WF_JSD_to_JAGP/fort.10 .
cp ../01convert_WF_JSD_to_JAGP/fort.10_in fort.10_corr
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_read;
cat corrsampling.dat
03-03 Li2 dimer and Li atom: VMC-optimization (WF=JsAGPs)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
Warning
If you want to optimized the contraction coefficients, you should put
iesup=1 in the ¶meters section. The code optimizes also
contraction coefficients when you put iesup=1 with itestr4=-4
or itestr4=-9. When you put iesup=1 with itestr4=-8
or itestr4=-5, the code optimizes not only contraction coefficients
but also exponents of basis set.
#copy wavefunction
cd ../03optimization/
cp ../01convert_WF_JSD_to_JAGP/fort.10 .
cp fort.10 fort.10_jsd
# datasmin.input
...
¶meters
iesd=1
iesfree=1
iessw=1
iesup=1
...
# commands
mpirun -np 4 turborvb-mpi.x < datasmin.input > out_min;
readalles.x
readalles.x << ____EOS
1 81 1 0
0
1000
____EOS
03-04 Li2 dimer and Li atom: VMC (WF=JsAGPs)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../04vmc
cp ../03optimization/fort.10 .
cp ../03optimization/datasmin.input ave.in
gsed -i -e 's/ngen=.*/ngen=1/g' ave.in
gsed -i -e 's/iopt=.*/iopt=1/g' ave.in
line_num_key=`grep "unconstrained" -n fort.10 | cut -d ":" -f 1`
line_num=`expr $line_num_key + 1`
line=`gsed -n ${line_num}p fort.10`
mod_line=`echo $line | gsed -e 's/0$/ 1/'`
gsed -i "${line_num}d" fort.10
gsed -i "${line_num_key}a $mod_line" fort.10
turborvb-serial.x < ave.in > out_ave
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
forcevmc.sh 10 2 1
cat pip0.d | grep "Energy"
03-05 Li2 dimer and Li atom: LRDMC (WF=JsAGPs)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../05lrdmc/
cp ../04vmc/fort.10 .
alat_list="0.8Z 1.0Z 1.2Z 1.5Z"
lrdmc_root_dir=`pwd`
for alat in $alat_list
do
cd alat_${alat}
cp ${lrdmc_root_dir}/fort.10 ./fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
mpirun -np 4 turborvb-mpi.x < datasfn.input > out_fn;
cd ${lrdmc_root_dir}
done
num=0
echo -n > ${lrdmc_root_dir}/evsa.gnu
for alat in $alat_list
do
cd alat_${alat}
num=`expr ${num} + 1`
echo "10 20 5 1" | readf.x
alat_d=`grep alat= datasfn.input | cut -f 2 -d '='`
echo -n "${alat_d} " >> ${lrdmc_root_dir}/evsa.gnu
tail -n 1 fort.20 | awk '{print $1, $2}' >> ${lrdmc_root_dir}/evsa.gnu
cd ${lrdmc_root_dir}
done
gsed "1i 2 ${num} 4 1" evsa.gnu > evsa.in
funvsa.x < evsa.in > evsa.out
cat evsa.out
Finally I got:
Li2 dimer
HF = -14.8715 Ha
VMC(JDFT) = -14.9759(9) Ha
LRDMC(JDFT) = -14.9894(11) Ha
VMC(JSD) = -14.9803(5) Ha
LRDMC(JSD) = -14.9909(12) Ha
VMC(JsAGPs) = -14.9821(4) Ha
LRDMC(JsAGPs) = -14.991(1) Ha
Exact = -14.9951 Ha
Li atom
HF = -7.4327 Ha
VMC(JDFT) = -7.4754(3) Ha
LRDMC(JDFT) = -7.4779(5) Ha
VMC(JSD) = -7.4769(2) Ha
LRDMC(JSD) = -7.4779(4) Ha
VMC(JsAGPs) = -7.477(2) Ha
LRDMC(JsAGPs) = -7.4783(4) Ha
Exact = -7.4781 Ha
Binding energy
Ebond = 38.9 mHa (experiment)
Warning
For a real run (i.e., for a peer-reviewed paper), one should optimize variational parameters much more carefully. We recommend that one consult to an expert or a developer of TurboRVB, or carefully read the 98 Wavefuntion optimization part.
04 Li2 dimer and Li atom - JAGPu ansatz¶
04-01 Li2 dimer: prepration of a wave function¶
As in the JDFT procedure, prepare Li2 dimer structure,
Li2.xyz
2
comment line
Li 0.0000 0.0000 -2.52589975576638451762
Li 0.0000 0.0000 2.52589975576638451762
The point is that you should set twobody=-22 and symmagp=.false. in makefort10.input
# makefort10.input
&system
posunits='bohr'
natoms=2
ntyp=1
pbcfort10=.false.
/
&electrons
twobody=-22
twobodypar=1.00
onebodypar=0.90
no_4body_jas=.false.
neldiff=0
/
&symmetries
eqatoms=.true.
rot_det=.true.
symmagp=.false.
/
ATOMIC_POSITIONS
3.0000000000000000 3.0000000000000000 0.0000000000000000 0.0000000000000000 -2.5259024244810400
3.0000000000000000 3.0000000000000000 0.0000000000000000 0.0000000000000000 2.5259024244810400
/
ATOM_3
&shells
nshelldet=11
nshelljas=5
/
1 1 16
1 14.24
1 1 16
1 4.581
1 1 16
1 1.58
1 1 16
1 0.564
1 1 16
1 0.07345
1 1 16
1 0.02805
1 1 36
1 1.534
1 1 36
1 0.2749
1 1 36
1 0.07362
1 1 36
1 0.02403
1 1 68
1 0.1144
# Parameters atomic Jastrow wf
1 1 16
1 4.581
1 1 16
1 1.58
1 1 16
1 0.564
1 1 36
1 0.2749
1 1 36
1 0.07362
# convertfort10mol.input
&control
epsdgm=-1d-14
/
&mesh_info
ax=0.20
nx=64
ny=64
nz=128
/
&molec_info
nmol=3
nmolmax=3
nmolmin=3
/
The other procedure is the same. You may obtain makefort10.input. Next, you can run makefort10.sh:
kosukenoMBP% ./makefort10.sh
where,
# makefort10.sh
kosukenoMBP% cat makefort10.sh
makefort10.x < makefort10.input > out_make
mv fort.10_new fort.10
mv fort.10 fort.10_in
convertfort10mol.x < convertfort10mol.input > out_mol
mv fort.10_new fort.10
The generated wave function fort.10 is a Jastrow Antisymmetrized Geminal Power including a triplet correlation (JAGPu).
Warning
If you want to use a more general spin-dependent Jastrow, i.e., independent parameters for the parallel and opposite spins (twobody = -27), you should manually put variational parameters for the two-body Jastrow part after generating fort.10. Since makefort10.x supports only one variable for the two-body part at present, while -27 has two independent two-body Jastrow variational parameters.
Indeed, If you want to use a more general spin-dependent Jastrow, whenever you generate a new fort.10 file, you should replace
# Parameters Jastrow two body
-2 1.00000000000000 0.900000000000000
in a generated fort.10 with
# Parameters Jastrow two body
-3 1.00000000000000 1.00000000000000 0.900000000000000
If the number of species in a system is more than 1, e.g., LiH, you should also put independent one-body Jastrows. Namely, you should replace
# Parameters Jastrow two body
-2 1.00000000000000 0.900000000000000
in a generated fort.10 with
# Parameters Jastrow two body
-4 1.00000000000000 1.00000000000000 0.900000000000000 0.900000000000000
04-02 Li2 dimer: generate a trial wave function using DFT.¶
The next step is to generate a trial wave function using the built-in DFT code. This is the minimal input file:
# prep.input
&simulation
itestr4=-4
iopt=1
double_mesh=.true
/
&pseudo
/
&vmc
/
&optimization
molopt=1
/
&readio
/
¶meters
/
&molecul
ax=0.2
ay=0.2
az=0.2
nx=64
ny=64
nz=128
/
&dft
maxit=50
epsdft=1d-5
mixing=1.0d0
typedft=4
optocc=1 ! do not fix the occ.
l0_at=1.0
scale_z=5
scale_hartree=-1.00
corr_hartree=.true.
linear=.false.
nxs=1
nys=1
nzs=2
h_field=1.0d-2
/
1 -1 ! <- magnetic moment (i.e., * h_field)
Then, you can run DFT by typing:
mpirun -np 4 prep-mpi.x < prep.input > out_prep
04-03 Li2 dimer: Check local magnetic moments¶
You can check the obtained magnetic moments by using plot_orbitals.x
# copy fort.10
KosukenoMacBook-Pro-2% cp ../01DFT/fort.10_new fort.10
# plot spin density
KosukenoMacBook-Pro-2% plot_orbitals.x
Number of molecular orbitals : 6
Choose box size (x,y,z)
15 15 15
15.0000000000000 15.0000000000000 15.0000000000000
Choose number of mesh points (x,y,z) :
61 61 61
61 61 61
Choose orbitals to tabulate (possible answers all, partial, charge, spin) :
spin
spin
Please give the lowest molecular orbital within 1 and 6
1
Number of fully occupied molecular orbital/total number occupied by up and down?
3 3
Momentum magnetization ? (unit 2pi/cellscale)
0 0 0
You may obtain --xsf output_spin000000.xsf. Depict it using xcrysden:
# KosukenoMacBook-Pro-2% xcrysden --xsf output_spin000000.xsf
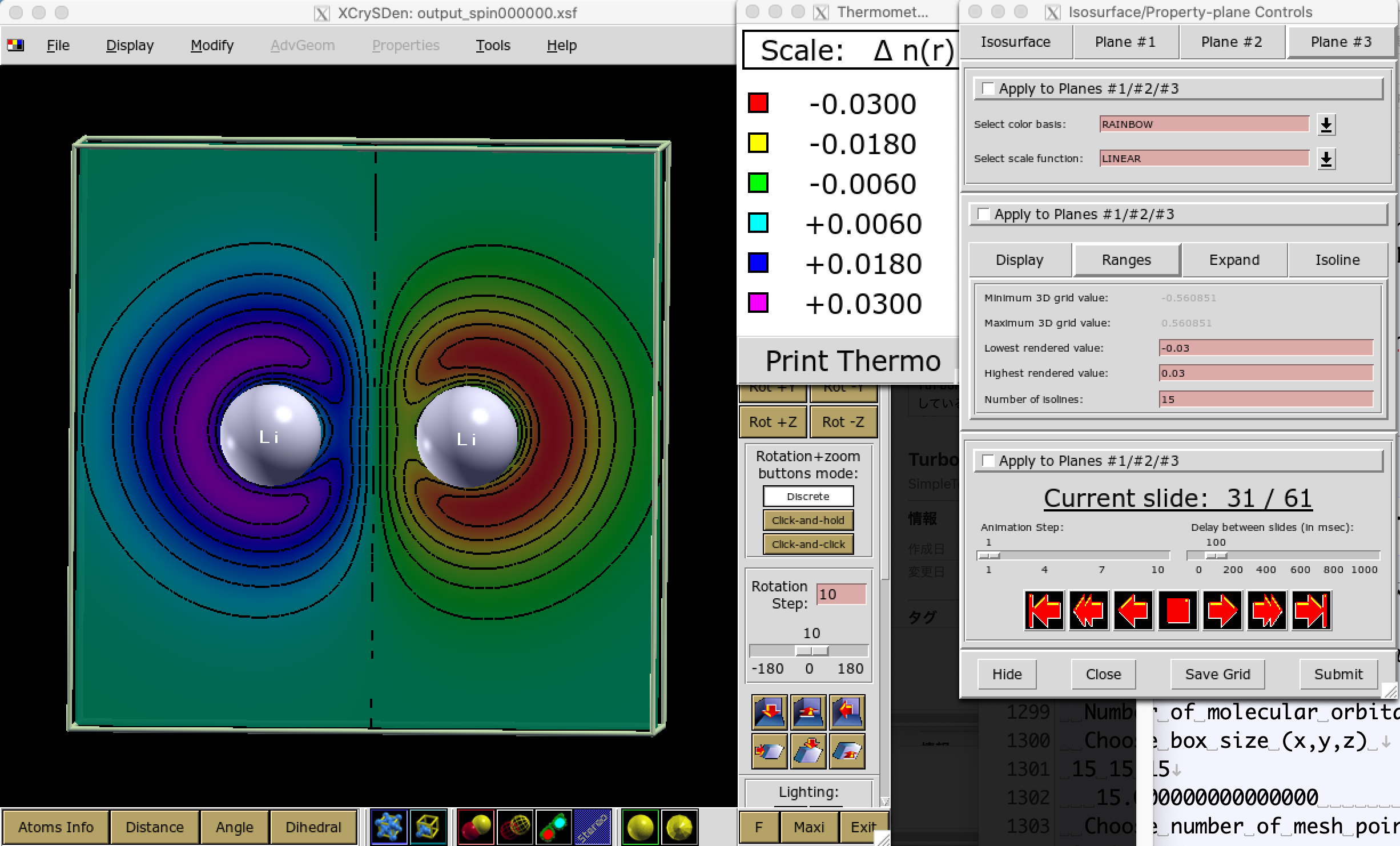
Thus, one can obtain an AFM trial wavefunction.
04-04 Li2 dimer and Li atom: Convert JDFT WF to JAGPu one¶
Next step is to convert the optimized JDFT ansatz to a JAGPu one. You should check the consistency after conversion.
# conversion of WF (02convert_WF_JDFT_to_JAGP)
cp ../01trial_wavefunction/01DFT/fort.10_new ./fort.10_in
cp ../01trial_wavefunction/00makefort10/makefort10.input .
sym_line=`grep "&symmetries" -n makefort10.input | cut -d ":" -f 1`
sym_line=`expr $sym_line + 1`
gsed -i "${sym_line}i nosym_contr=.true." makefort10.input
hyb_line=`grep "nshelljas" -n makefort10.input | cut -d ":" -f 1`
hyb_line=`expr $hyb_line + 1`
gsed -i "${hyb_line}i ndet_hyb=2" makefort10.input
makefort10.x < makefort10.input > out_make
mv fort.10_new fort.10_out
convertfort10.x < convertfort10.input > out_conv
mv fort.10_new fort.10
# correlated sampling (03conversion_check)
cp ../02convert_WF_JDFT_to_JAGP/fort.10 .
cp ../02convert_WF_JDFT_to_JAGP/fort.10_in fort.10_corr
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_read
cat corrsampling.dat
# Li-dimer
Number of bins 146
reference energy: E(fort.10) -0.149056547E+02 0.903277814E-02
reweighted energy: E(fort.10_corr) -0.149153980E+02 0.890288018E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.974326537E-02 0.795833755E-03
Overlap square : (fort.10,fort.10_corr) 0.993230204E+00 0.284523743E-03
# Li-atom
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468673E-02
reweighted energy: E(fort.10_corr) -0.148896681E+02 0.929468634E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.124625377E-07 0.316227766E-07
Overlap square : (fort.10,fort.10_corr) 0.999999997E+00 0.316227766E-07
04-05 Li2 dimer and Li atom: Optimization (WF=JAGPu)¶
Now that you have obtained a good trial JAGPu wavefunction, you can optimize its nodal surface at the VMC level.
Then, copy the converted WF.
cp ../02convert_WF_JDFT_to_JAGP/fort.10 ./
cp fort.10 fort.10_dft
Prepare datasmin.input:
&simulation
itestr4=-4
ngen=200000
iopt=1
maxtime=10800
!nscra=1
/
&pseudo
/
&vmc
/
&optimization
ncg=2
nweight=1000
nbinr=10
iboot=0
tpar=1.5d-1
parr=5.0d-3
/
&readio
!iread=3
!nowrite12=.true.
/
¶meters
iesd=1
iesfree=1
iessw=1
/
The difference from datasmin.input in 03optimization is iessw,
because we optimize the determinant part (nodal surface) at this step.
Run a VMC-opt run.
#run vmc-opt
mpirun -np 4 turborvb-mpi.x < datasmin.input > out_min
#average fort.10
readalles.x << ____EOS
1 81 1 0
0
1000
____EOS
The rest of the procesure (e.g., averaging the variational parameters) is the same.
Warning
For a real run (i.e., for a peer-reviewed paper), one should optimize variational parameters much more carefully. We recommend that one consult to an expert user or a developer of TurboRVB, or carefully read the 98 Wavefuntion optimization part.
04-06 Li2 dimer and Li atom: VMC and LRDMC¶
VMC and LRDMC procesures are the same as in the JsAGPs case.
# VMC
cp ../04optimization/fort.10 ./
cp ../04optimization/datasmin.input ave.in
gsed -i -e 's/ngen=.*/ngen=1/g' ave.in
gsed -i -e 's/iopt=.*/iopt=1/g' ave.in
line_num_key=`grep "unconstrained" -n fort.10 | cut -d ":" -f 1`
line_num=`expr $line_num_key + 1`
line=`gsed -n ${line_num}p fort.10`
mod_line=`echo $line | gsed -e 's/0$/ 1/'`
gsed -i "${line_num}d" fort.10
gsed -i "${line_num_key}a $mod_line" fort.10
turborvb-serial.x < ave.in > out_ave
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
forcevmc.sh 20 5 1
cat pip0.d | grep "Energy"
#LRDMC
cp ../05vmc/fort.10 .
alat_list="0.8Z 1.0Z 1.2Z 1.5Z"
lrdmc_root_dir=`pwd`
for alat in $alat_list
do
cd alat_${alat}
cp ${lrdmc_root_dir}/fort.10 ./fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
mpirun -np 4 turborvb-mpi.x < datasfn.input > out_fn;
cd ${lrdmc_root_dir}
done
num=0
echo -n > ${lrdmc_root_dir}/evsa.gnu
for alat in $alat_list
do
cd alat_${alat}
num=`expr ${num} + 1`
echo "10 20 5 1" | readf.x
alat_d=`grep alat= datasfn.input | cut -f 2 -d '='`
echo -n "${alat_d} " >> ${lrdmc_root_dir}/evsa.gnu
tail -n 1 fort.20 | awk '{print $1, $2}' >> ${lrdmc_root_dir}/evsa.gnu
cd ${lrdmc_root_dir}
done
gsed "1i 2 ${num} 4 1" evsa.gnu > evsa.in
funvsa.x < evsa.in > evsa.out
Li2 dimer
HF = -14.8715 Ha
VMC(JDFT) = -14.9759(9) Ha
LRDMC(JDFT) = -14.9894(11) Ha
VMC(JsAGPs) = -14.9821(4) Ha
LRDMC(JsAGPs) = -14.991(1) Ha
VMC(JAGPu) = -14.9819(5) Ha
LRDMC(JAGPu) = -14.990(1) Ha
Exact = -14.9951 Ha
Li atom
HF = -7.4327 Ha
VMC(JDFT) = -7.4754(3) Ha
LRDMC(JDFT) = -7.4779(5) Ha
VMC(JsAGPs) = -7.477(2) Ha
LRDMC(JsAGPs) = -7.4783(4) Ha
VMC(JAGPu) = -7.477(3) Ha
LRDMC(JAGPu) = -7.4791(4) Ha
Exact = -7.4781 Ha
Binding energy
Ebond = 38.9 mHa (experiment)
05 Li2 dimer and Li atom - JAGP (JPf) ansatz¶
05-01 (spin-unpolarized case) Li2 dimer: Convert JDFT WF to JAGP one¶
The most important procedure in a Pfaffian calculation is to convert a JDFT or JAGPu ansatz to JAGP(JPf) ansatz. Since the JAGP ansatz is a special case of the JPf one, where only G_{ud} and G_{du} terms are defined as described in the section review paper, the conversion can be realized just by direct substitution. Therefore, the main challenge is to find a reasonable initialization for the two spin-triplet sectors G_{uu} and G_{dd} that are not described in the JAGP and that otherwise have to be set to 0. There are two possible approaches to convert an ansatz: \rm(\hspace{.18em}i\hspace{.18em}) for polarized systems, we can build the G_{uu} block of the matrix by using an even number of \{ \phi_i\} and build an antisymmetric g_{uu}, where the eigenvalues \lambda_k are chosen to be large enough to occupy certainly these unpaired states, as in the standard Slater determinant used for our initialization. Again, we emphasize that this works only for polarized systems. \rm(\hspace{.08em}ii\hspace{.08em}) The second approach that also works in a spin-unpolarized case is to determine a standard broken symmetry single determinant ansatz ({it e.g.}, by TurboRVB built-in DFT within the LSDA) and modify it with a global spin rotation. Indeed, in the presence of finite local magnetic moments, it is often convenient to rotate the spin moments of the WF in a direction perpendicular to the spin quantization axis chosen for our spin-dependent Jastrow factor, {it i.e.}, the z quantization axis. In this way one can obtain reasonable initializations for G_{uu} and G_{dd}. TurboRVB allows every possible rotation, including an arbitrary small one close to the identity. A particularly important case is when a rotation of \pi/2 is applied around the y direction. This operation maps |\uparrow \rangle \rightarrow \frac{1} {\sqrt{2}} \left( | \uparrow \rangle + |\downarrow \rangle \right) \mbox{ and } |\downarrow \rangle \rightarrow \frac 1 {\sqrt{2}} \left( | \uparrow \rangle - |\downarrow \rangle \right). One can convert from a AGP the pairing function that is obtained from a VMC optimization {g_{ud}}(\mathbf{i},\mathbf{j}) = {f_S}({{\mathbf{r}}_i},{{\mathbf{r}}_j})\frac{{\left| { \uparrow \downarrow } \right\rangle - \left| { \downarrow \uparrow } \right\rangle }}{{\sqrt 2 }} + {f_T}({{\mathbf{r}}_i},{{\mathbf{r}}_j})\frac{{\left| { \uparrow \downarrow } \right\rangle + \left| { \downarrow \uparrow } \right\rangle }}{{\sqrt 2 }} to a Pf one {g_{ud}}(\mathbf{i},\mathbf{j}) \to g\left( {\mathbf{i},\mathbf{j}} \right){\text{ }} = {f_S}({{\mathbf{r}}_i},{{\mathbf{r}}_j})\frac{{\left| { \uparrow \downarrow } \right\rangle - \left| { \downarrow \uparrow } \right\rangle }}{{\sqrt 2 }} + {f_T}({{\mathbf{r}}_i},{{\mathbf{r}}_j})\left( {\left| { \uparrow \uparrow } \right\rangle - \left| { \downarrow \downarrow } \right\rangle } \right). This transformation provides a meaningful initialization to the Pfaffian WF that can be then optimized for reaching the best possible description of the ground state within this ansatz.
The strategy \rm(\hspace{.08em}ii\hspace{.08em}) is employed for the Li dimer (i.e., unpolarized case) while \rm(\hspace{.18em}i\hspace{.18em}) is employed for the Li atom (i.e., polarized case)
################
# Li-dimer, strategy (ii)
################
# run VMC for corr. sampling.
cp ../../../04JAGPu/01Li2_dimer/01trial_wavefunction/01DFT/fort.10_new fort.10_in
cp fort.10_in fort.10_dft
cp fort.10_dft fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc
rm fort.10
################
# convert JDFT -> uncont JAGP
################
# convertfort.10
cp ../../../04JAGPu/01Li2_dimer/01trial_wavefunction/00makefort10/makefort10.input makefort10_agp_uncont.input
makefort10.x < makefort10_agp_uncont.input > out_make_agp_uncont
mv fort.10_new fort.10_out
convertfort10.x < convertfort10.input > out_conv_agp_uncont
mv fort.10_new fort.10_agp_uncont
# corr sampling
cp fort.10_dft fort.10
cp fort.10_agp_uncont fort.10_corr
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_dft_agp_uncont.dat
rm fort.10
cat corrsampling_dft_agp_uncont.dat
KosukenoMacBook-Pro-2% cat corrsampling_dft_agp_uncont.dat
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468674E-02
reweighted energy: E(fort.10_corr) -0.148896681E+02 0.929468647E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.122986368E-07 0.316227766E-07
Overlap square : (fort.10,fort.10_corr) 0.999999997E+00 0.316227766E-07
################
# convert uncont JAGP -> uncont JPf (norotate)
################
cp fort.10_agp_uncont fort.10_in
cp makefort10_agp_uncont.input makefort10_pf_uncont.input
vi makefort10_pf_uncont.input
&system
...
yes_pfaff=.true.
&symmetries
...
rot_pfaff=.false.
makefort10.x < makefort10_pf_uncont.input > out_make_pfaff_uncont
mv fort.10_new fort.10_out
convertpfaff.x norotate
cp fort.10_new fort.10_pfaff_uncont
# corr. sampling
cp fort.10_dft fort.10
cp fort.10_pfaff_uncont fort.10_corr
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_agp_pfaff_uncont.dat
cat corrsampling_agp_pfaff_uncont.dat
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468674E-02
reweighted energy: E(fort.10_corr) -0.148896681E+02 0.929468647E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.122986368E-07 0.316227766E-07
Overlap square : (fort.10,fort.10_corr) 0.999999997E+00 0.316227766E-07
################
# convert uncont JPf -> cont JPf
################
cp fort.10_pfaff_uncont fort.10_in
cp makefort10_pf_uncont.input makefort10_pf_cont.input
vi makefort10_pf_cont.input
&symmetries
nosym_contr=.true.
ATOM_3
&shells
nshelldet=11
nshelljas=5
ndet_hyb=2
makefort10.x < makefort10_pf_cont.input > out_make_pfaff_cont
mv fort.10_new fort.10_out
convertfort10.x < convertfort10.input > out_conv_pfaff_cont
cp fort.10_new fort.10_pfaff_cont
cp fort.10_pfaff_cont fort.10_corr
# corr. sampling
cp fort.10_dft fort.10
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_dft_pfaff_cont.dat
cat corrsampling_dft_pfaff_cont.dat
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468674E-02
reweighted energy: E(fort.10_corr) -0.148896681E+02 0.929468645E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.115194734E-07 0.316227766E-07
Overlap square : (fort.10,fort.10_corr) 0.999999997E+00 0.505322962E-07
################
# rotate fort.10
################
# rotate by 1/8pi
cp fort.10_pfaff_cont fort.10_in
cp fort.10_pfaff_cont fort.10_out
convertpfaff.x
-0.125
cp fort.10_new fort.10_pfaff_cont_rot
# clean fort.10
cp fort.10_pfaff_cont_rot fort.10
cleanfort10.x
mv fort.10_clean fort.10
cp fort.10 fort.10_final
cp fort.10 fort.10_corr
# corr. sampling
cp fort.10_dft fort.10
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_dft_pfaff_cont_rot.dat
rm fort.10
cat corrsampling_dft_pfaff_cont_rot.dat
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468674E-02
reweighted energy: E(fort.10_corr) -0.148904372E+02 0.936290264E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.769116534E-03 0.396919093E-03
Overlap square : (fort.10,fort.10_corr) 0.998728350E+00 0.512541253E-03
The total energy was a bit lost by the rotation.
Warning
If you want to use a more general spin-dependent Jastrow, i.e., independent parameters for the parallel and opposite spins (twobody = -27), you should manually put variational parameters for the two-body Jastrow part whenever generating a new fort.10.
05-02 (spin-polarized case) Li atom: Convert JDFT WF to JAGP one¶
################
# Li-atom, strategy (i)
################
# run VMC for corr. sampling.
cp ../../../04JAGPu/02Li_atom/01trial_wavefunction/01DFT/fort.10_new fort.10_in
cp fort.10_in fort.10_dft
cp fort.10_dft fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc
rm fort.10
################
# convert JDFT -> uncont JAGP
################
# convertfort.10
cp ../../../04JAGPu/02Li_atom/01trial_wavefunction/00makefort10/makefort10.input makefort10_agp_uncont.input
makefort10.x < makefort10_agp_uncont.input > out_make_agp_uncont
mv fort.10_new fort.10_out
convertfort10.x < convertfort10.input > out_conv_agp_uncont
mv fort.10_new fort.10_agp_uncont
# corr sampling
cp fort.10_dft fort.10
cp fort.10_agp_uncont fort.10_corr
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_dft_agp_uncont.dat
rm fort.10
cat corrsampling_dft_agp_uncont.dat
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468674E-02
reweighted energy: E(fort.10_corr) -0.148896681E+02 0.929468647E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.122986368E-07 0.316227766E-07
Overlap square : (fort.10,fort.10_corr) 0.999999997E+00 0.316227766E-07
################
# convert uncont JAGP -> uncont JPf (norotate)
################
cp fort.10_agp_uncont fort.10_in
cp makefort10_agp_uncont.input makefort10_pf_uncont.input
# makefort10_pf_uncont.input
&system
...
yes_pfaff=.true.
&symmetries
...
rot_pfaff=.false.
makefort10.x < makefort10_pf_uncont.input > out_make_pfaff_uncont
mv fort.10_new fort.10_out
convertpfaff.x norotate
10000
cp fort.10_new fort.10_pfaff_uncont
# corr. sampling
cp fort.10_dft fort.10
cp fort.10_pfaff_uncont fort.10_corr
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_agp_pfaff_uncont.dat
cat corrsampling_agp_pfaff_uncont.dat
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468674E-02
reweighted energy: E(fort.10_corr) -0.148896681E+02 0.929468647E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.122986368E-07 0.316227766E-07
Overlap square : (fort.10,fort.10_corr) 0.999999997E+00 0.316227766E-07
################
# convert uncont JPf -> cont JPf
################
cp fort.10_pfaff_uncont fort.10_in
cp makefort10_pf_uncont.input makefort10_pf_cont.input
# makefort10_pf_cont.input
&symmetries
nosym_contr=.true.
ATOM_3
&shells
nshelldet=11
nshelljas=5
ndet_hyb=2
makefort10.x < makefort10_pf_cont.input > out_make_pfaff_cont
mv fort.10_new fort.10_out
convertfort10.x < convertfort10.input > out_conv_pfaff_cont
cp fort.10_new fort.10_pfaff_cont
# clean fort.10
cp fort.10_pfaff_cont fort.10
cleanfort10.x
mv fort.10_clean fort.10
cp fort.10 fort.10_final
cp fort.10 fort.10_corr
# corr. sampling
cp fort.10_dft fort.10
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_dft_pfaff_cont.dat
cat corrsampling_dft_pfaff_cont.dat
Number of bins 146
reference energy: E(fort.10) -0.148896680E+02 0.929468674E-02
reweighted energy: E(fort.10_corr) -0.148896681E+02 0.929468645E-02
reweighted difference: E(fort.10)-E(fort.10_corr) 0.115194734E-07 0.316227766E-07
Overlap square : (fort.10,fort.10_corr) 0.999999997E+00 0.505322962E-07
05-03 Li2 dimer and Li atom: VMC optimization (WF=JPf)¶
# Li2-dimer
cp ../01convert_WF_JDFT_to_JAGP/fort.10_pfaff_cont_rot fort.10
cp fort.10 fort.10_sav
cp ../01convert_WF_JDFT_to_JAGP/makefort10_pf_cont.input .
# Li-atom
cp ../01convert_WF_JDFT_to_JAGP/fort.10_pfaff_cont fort.10
cp fort.10 fort.10_sav
cp ../01convert_WF_JDFT_to_JAGP/makefort10_pf_cont.input .
# Li2-dimer and Li-atom
mpirun -np 4 turborvb-mpi.x < datasmin.input > out_min &
cleanfort10.x
mv fort.10_clean fort.10
# check convergence
readalles.x << ____EOS
1 1 0 1
0
1000
____EOS
# average
readalles.x << ____EOS
1 81 1 0
0
1000
____EOS
!! If you find many War in your output during optimization, !!
!! please do the following stabilization, and continue optimization. !!
# Stabilization (especially needed for an open system, Li atom)
cp fort.10 fort.10_in
cp fort.10 fort.10_opt
vi convertfort10mol.input
# you should comment out epsdgm=-1d-14 -> !epsdgm=1d-14.
# If this value is set negative, a generated molecular orbitals are random.
# You can start from nmolmin=1, until nmolmin=N_el/ 2.
# and check you do not loose much energy.
convertfort10mol.x < convertfort10mol.input > out_mol_stabilized
cp fort.10_new fort.10
# Correlated sampling (after the above conversion)
cp fort.10_in fort.10_corr
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc
mpirun -np 4 readforward-mpi.x < datasvmc.input > out_readforward
mv corrsampling.dat corrsampling_pfaff_stabilized.dat
cat corrsampling_pfaff_stabilized.dat
# Convert back to a geminal (JAGP)
cp ../01convert_WF_JDFT_to_JAGP/makefort10_pf_cont.input .
cp fort.10 fort.10_in
twobodyjas=`grep -A 1 "Parameters Jastrow two body" ./fort.10_in | tail -n -1 | awk '{print $2}'`
onebodyjas=`grep -A 1 "Parameters Jastrow two body" ./fort.10_in | tail -n -1 | awk '{print $3}'`
echo "twobodyjas=${twobodyjas} onebodyjas=${onebodyjas}"
gsed -i -e "/twobodypar/c\ twobodypar=${twobodyjas}" \
-e "/onebodypar/c\ onebodypar=${onebodyjas}" \
makefort10_pf_cont.input
makefort10.x < makefort10_pf_cont.input > out_make_pfaff_cont
mv fort.10_new fort.10_out
convertfort10.x < convertfort10.input > out_conv
cp fort.10_new fort.10
cleanfort10.x
mv fort.10_clean fort.10
cp fort.10_opt fort.10_new
copyjas.x
Warning
If you are using a more general spin-dependent Jastrow (i.e., twobody = -27), you should manually put Jastrow parameters after generating fort.10_out by makerfort10.x.
05-04 Li2 dimer and Li atom: VMC (WF=JPf)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../03vmc
cp ../02optimization/fort.10 .
cp ../02optimization/datasmin.input ave.in
gsed -i -e 's/ngen=.*/ngen=1/g' ave.in
gsed -i -e 's/iopt=.*/iopt=1/g' ave.in
line_num_key=`grep "unconstrained" -n fort.10 | cut -d ":" -f 1`
line_num=`expr $line_num_key + 1`
line=`gsed -n ${line_num}p fort.10`
mod_line=`echo $line | gsed -e 's/0$/ 1/'`
gsed -i "${line_num}d" fort.10
gsed -i "${line_num_key}a $mod_line" fort.10
turborvb-serial.x < ave.in > out_ave
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
forcevmc.sh 10 2 1
cat pip0.d | grep "Energy"
05-05 Li2 dimer and Li atom: LRDMC (WF=JPf)¶
Please refer to the Hydrogen tutorial for the details. Here, only needed commands are shown.
cd ../04lrdmc/
cp ../03vmc/fort.10 .
alat_list="0.8Z 1.0Z 1.2Z 1.5Z"
lrdmc_root_dir=`pwd`
for alat in $alat_list
do
cd alat_${alat}
cp ${lrdmc_root_dir}/fort.10 ./fort.10
mpirun -np 4 turborvb-mpi.x < datasvmc.input > out_vmc;
mpirun -np 4 turborvb-mpi.x < datasfn.input > out_fn;
cd ${lrdmc_root_dir}
done
num=0
echo -n > ${lrdmc_root_dir}/evsa.gnu
for alat in $alat_list
do
cd alat_${alat}
num=`expr ${num} + 1`
echo "10 20 5 1" | readf.x
alat_d=`grep alat= datasfn.input | cut -f 2 -d '='`
echo -n "${alat_d} " >> ${lrdmc_root_dir}/evsa.gnu
tail -n 1 fort.20 | awk '{print $1, $2}' >> ${lrdmc_root_dir}/evsa.gnu
cd ${lrdmc_root_dir}
done
gsed "1i 2 ${num} 4 1" evsa.gnu > evsa.in
funvsa.x < evsa.in > evsa.out
Finally I got:
Li2 dimer
HF = -14.8715 Ha
VMC(JDFT) = -14.9759(9) Ha
LRDMC(JDFT) = -14.989(1) Ha
VMC(JsAGPs) = -14.9821(4) Ha
LRDMC(JsAGPs) = -14.991(1) Ha
VMC(JAGPu) = -14.9819(5) Ha
LRDMC(JAGPu) = -14.990(1) Ha
VMC(JAGP) = -14.9831(4) Ha
LRDMC(JAGP) = -14.993(1) Ha
Exact = -14.9951 Ha
Li atom
HF = -7.4327 Ha
VMC(JDFT) = -7.4754(3) Ha
LRDMC(JDFT) = -7.4779(5) Ha
VMC(JsAGPs) = -7.477(2) Ha
LRDMC(JsAGPs) = -7.4783(4) Ha
VMC(JAGPu) = -7.477(3) Ha
LRDMC(JAGPu) = -7.4791(4) Ha
VMC(JAGP) = -7.4766(3) Ha
LRDMC(JAGP) = -7.4784(4) Ha
Exact = -7.4781 Ha
Binding energy
Ebond = 38.9 mHa (experiment)